-
Để giúp anh/chị
quyết định có đọc tiếp hay không, tôi xin phép cung cấp các thông tin liên quan
đến bài post này như sau:
·
Chủ
đề: Machine Learning,
Bioinformatics, Protein
·
Tính
thời sự: Tháng 10/2024.
·
Thời
gian đọc: 8 phút, kể cả
thời gian uống cà phê.
-
Giải Nobel năm
nay khá đặc biệt: giải Nobel Vật lý và giải Nobel Hóa học đều trao cho những
người làm về CNTT, là các nhà khoa học về lĩnh vực Machine Learning.
·
Giải
Nobel Vật lý trao cho ông John J. Hopfield – giáo sư tại ĐH Princeton,
Hoa Kỳ và cho ông Geoffrey E. Hinton – giáo sư tại ĐH Toronto, Canada.
·
Giải
Nobel Hóa học, một nửa trao cho David Baker – ĐH Washington, Hoa Kỳ, một
nửa trao cho Demis Hassabis và John M. Jumper – công ty Google
DeepMind, London, Vương quốc Anh.
Để phục vụ
anh/chị nhâm nhi cà phê lần này, tôi xin phép đàm luận về protein (đạm) – là chủ
đề của giải Nobel Hóa học năm nay (2024). Protein là thành phần cơ bản của sự sống
(life). Dưới góc độ hệ thống điện toán, một số protein đảm nhận chức
năng cảm biến, một số khác đảm nhận chức năng thực thi, một số lại
đảm nhận chức năng quản trị. Nói một cách tổng quan: protein “vận hành”
sự sống.
Giải Nobel năm
nay gồm 2 nhánh: một nhánh sử dụng mạng nơ-ron (neural network) để phỏng
đoán (predict) cấu trúc 3D của protein từ một chuỗi axít amin cho trước
(Demis Hassabis và John M. Jumper). Nhánh còn lại là viết phần mềm
Rosetta rồi dùng phần mềm này để tạo ra các protein hoàn toàn mới chưa hề có
trong tự nhiên (David Baker).
⨭Về nhánh thứ nhất:
Tôi đã có dịp đàm
luận về vấn đề này trên diễn đàn ICT_VN vào ngày 30/07/2021
(cách đây hơn 3 năm). Đại ý: khi nghiên cứu protein, người ta nhận thấy có một
vấn đề rất thách thức, có tên gọi là “protein folding problem” (vấn đề cuộn gấp protein). Ông Christian B. Anfinsen, trong phát biểu nhận giải Nobel năm
1972, có nói, khi biết được trình tự chuỗi axít amin của một protein, về mặt
nguyên tắc, chúng ta sẽ biết cấu trúc 3D của protein đó. Định đề giả thuyết này
đã khơi mào cho một thách thức kéo dài suốt 5 thập kỷ (1972-2021). Đó là:
Câu đố: Cho biết cấu trúc một chiều (1D) của protein hãy tìm cấu
trúc ba chiều (3D) tương đương (duy nhất) của nó.
║ Chi tiết: Phát biểu trên không hẳn là đầy đủ, nó chỉ nói
lên được tính thách thức của vấn đề, chứ chưa đề cập đến tính đặc trưng của chuỗi
các axít amin. Định đề nói rằng, ở trong điều kiện môi trường nhất định (nhiệt
độ, nồng độ dung môi, v.v.) quá trình cuộn gấp xảy ra và chúng ta chú ý tính chất
này: cấu trúc nguyên bản (native structure) – sau khi quá trình cuộn gấp
kết thúc – là duy nhất. Nói cách khác, chỉ có duy nhất một cấu trúc 3D tương
đương với chuỗi trình tự ban đầu của protein – xem Anfinsen's dogma.
↓
·
Hãy
hình dung cấu trúc của một protein: giống như sợi các hạt gắn với nhau.
·
Các
hạt chính là các hóa chất có tên gọi là axit amin. Chỉ có 20 loại axít amin
khác nhau (Alanine, Arginine, Asparagine, Aspartic acid, Cysteine,
Glutamine, Glutamic acid, Glycine, Histidine, Isoleucine, Leucine, Lysine,
Methionine, Phenylalanine, Proline, Serine, Threonine, Tryptophan, Tyrosine,
Valine).
·
Các
sợi hạt này được lắp ráp, cuộn gấp tuân theo các “câu lệnh” (instruction)
nằm trong DNA (gen).
·
Lực
hút và lực đẩy giữa 20 loại axít amin khác nhau khiến chuỗi gấp lại theo kiểu
‘gấp giấy tự động’ trong nháy mắt, tạo thành các lọn, vòng và nếp gấp phức tạp,
đó chính là cấu trúc 3D của protein.
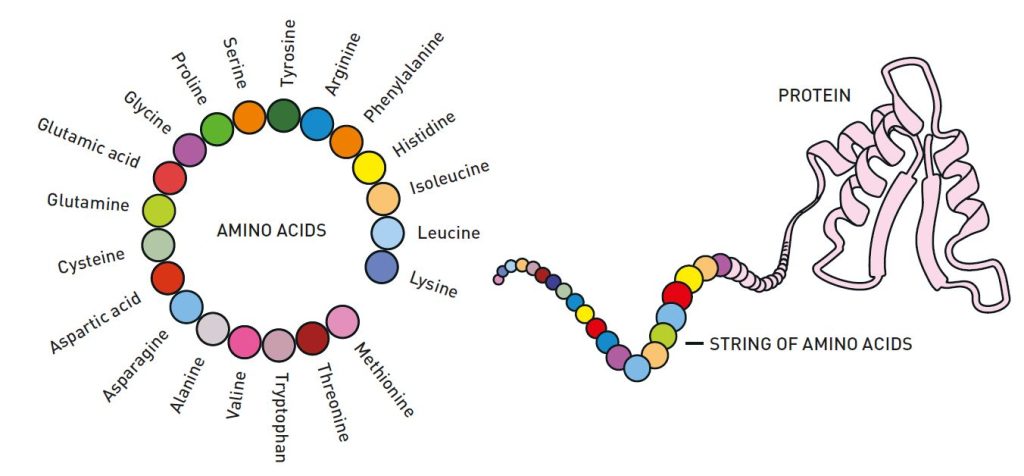 Chuỗi axit amin gấp lại thành cấu trúc ba chiều.
Hình ảnh minh họa cấu trúc của các axit amin và vai trò của chúng trong việc hình thành protein. Ở bên trái, có một vòng tròn hiển thị 20 loại axit amin khác nhau, đây là những khối xây dựng của protein. Các axit amin này bao gồm những loại quen thuộc như Glutamic acid, Lysine, Tyrosine, Methionine và những loại khác, mỗi loại được biểu diễn bằng một màu khác nhau. Bên phải của
sơ đồ vòng tròn, có một "chuỗi axit amin" tuyến tính, trực quan nối
các axit amin riêng lẻ trong một chuỗi giống như chuỗi dây chuyền. Chuỗi này
gấp lại thành một cấu trúc ba chiều phức tạp, đại diện cho một protein hoàn
chỉnh. Hình ảnh nhấn mạnh sự biến đổi từ một chuỗi đơn giản của các axit amin
thành một protein chức năng, có vai trò quan trọng trong các quá trình sinh học
khác nhau. |
Để xác định được
cấu trúc 3D của protein (xem hình ảnh minh họa), giới nghiên cứu đã sử dụng rất
nhiều phương pháp thí nghiệm hiện đại như nuclear magnetic resonance (cộng hưởng từ hạt nhân), X-ray crystallography (tinh thể học tia X), cryogenic electron microscopy (kính hiển vi điện tử đông lạnh). Tuy
đã sử dụng các công cụ hiện đại và đắt tiền (nhiều triệu đô-la một thiết bị)
nhưng việc làm thí nghiệm là một quá trình gian nan, mất nhiều công sức và phải
mất hàng tháng, hàng năm trời mới tìm ra được cấu trúc của một protein. Một cấu
trúc đã “vất vả” như thế, trong lúc ngoài tự nhiên có hàng triệu protein thì đến
bao giờ mới xong?!
Mở ngoặc ⦅
Bắt
đầu từ năm 1976, giới khoa học đã bắt đầu làm thí nghiệm để xác định cấu trúc
3D của protein (xem Protein structure determination). Tính đến tháng 4/2020, giới khoa học
đã xác định được 150,423 cấu trúc, trong đó chủ yếu bằng phương pháp tinh thể
học tia X (135,170), tiếp đến bằng phương pháp cộng hưởng từ hạt nhân
(11,337), bằng kính hiển vi điện tử đông lạnh (3,475), phương pháp
lai (155) và bằng các phương pháp khác (286) – xem Protein Data Bank (CSDL protein, viết tắt là PDB).
⚠ Chú
ý rằng đây là tập mẫu dữ liệu quan trọng và căn bản nếu phương
pháp dự đoán cấu trúc protein (Protein Structure Prediction) sử dụng mô
hình mạng nơ-ron (Neural Network).
Đóng ngoặc ⦆
Giải câu đố: Mô hình AlphaFold2.
Một cách tổng
quan:
[Chuỗi trình tự
1D] ⇨
{AlphaFold 2} ⇨ [Cấu trúc
3D (phỏng đoán)]
Nghĩa là: Cho đầu vào là chuỗi các axít amin, mô
hình {AlphaFold 2} cho đầu ra là cấu trúc 3D của protein (phỏng đoán). Cấu trúc
3D chính là tọa độ của các nguyên tử (heavy atom) của hạt tâm (axít amin).
Cấu trúc 3D (phỏng đoán) có độ chính xác đạt trên 90% so với kết quả bằng phương
pháp thí nghiệm.
Công việc trước đây mất nhiều năm giờ chỉ mất vài phút.
Khi Demis
Hassabis và John Jumper xác nhận AlphaFold2 thực sự hoạt động, họ đã tính toán
cấu trúc (3D) của tất cả các protein của con người. Sau đó, họ phỏng đoán cấu
trúc của hầu như tất cả 200 triệu protein mà các nhà nghiên cứu đã phát hiện ra
cho đến nay khi lập bản đồ các sinh vật trên Trái đất.
Google DeepMind
cũng đã công khai mã AlphaFold2 và bất kỳ ai cũng có thể truy cập. Mô hình AI
đã trở thành mỏ vàng cho các nhà nghiên cứu. Đến tháng 10 năm 2024, AlphaFold2
đã được hơn hai triệu người từ 190 quốc gia sử dụng. Trước đây, thường mất nhiều
năm để có được cấu trúc protein (nếu tìm ra). Bây giờ, có thể thực hiện trong
vài phút. Mô hình AI không hoàn hảo, nhưng nó ước tính độ chính xác của cấu
trúc mà nó tạo ra, do đó các nhà nghiên cứu biết mà dự liệu.
⨮
Về nhánh thứ hai:
Nhánh này nói về
lĩnh vực thiết kế protein - nghiên cứu tạo ra các protein “may đo theo yêu cầu”
– có tên gọi là de novo protein. Nghĩa là protein được thiết kế hoặc điều
chỉnh một cách đặc biệt để phù hợp với mục đích hoặc chức năng cụ
thể nào đó. Đây là trào lưu bắt đầu phát triển vào cuối những năm 1990. Trong
phần lớn các trường hợp, để tạo ra protein mới, người ta thường điều chỉnh các
protein đã có trong tự nhiên. Nhóm nghiên cứu của David Baker táo bạo hơn, đặt
vấn đề là tạo ra các protein hoàn toàn mới chưa hề có trong tự nhiên. Tất nhiên
là rất thách thức.
Vấn đề đầu tiên
là làm thế nào để thiết kế cấu trúc 3D của protein (sau khi đã cuộn gấp)
căn cứ theo mục đích hoặc chức năng. Sau khi có cấu trúc 3D rồi
thì câu hỏi tiếp theo là làm thế nào để biết chuỗi trình tự 1D tương đương của
cấu trúc 3D này. Và cuối cùng là bước kiểm thử: đưa protein được thiết kế “trên
giấy” vào “hiện trường” thật trong tự nhiên.
Sau khi thiết kế
cấu trúc 3D của protein, họ đưa thiết kế này làm đầu vào phần mềm Rosetta và đầu
ra là chuỗi axit amin (1D):
[Cấu trúc 3D (theo
thiết kế)] ⇨ {Rosetta}
⇨ [Chuỗi trình tự 1D]
Để xác thực, họ
đưa đoạn gen của chuỗi axit amin vào một loài vi khuẩn. Chúng ta hình dung là quá
trình cuộn gấp sẽ xảy ra một cách tự nhiên trong cơ thể của loài vi khuẩn đó. Sau
đó, họ xác định cấu trúc 3D của protein đã cuộn gấp trong vi khuẩn bằng phương
pháp tinh thể học tia X (X-ray crystallography). Protein mà họ tạo ra, có
tên gọi là “Top7”, có cấu trúc gần giống hệt như họ đã thiết kế.
Top7 là một tia sét bất ngờ đối với các nhà
nghiên cứu trong lĩnh vực thiết kế protein. Những người trước đây đã tạo ra
protein de novo chỉ có thể bắt chước các cấu trúc hiện có. Cấu trúc độc
đáo của Top7 không tồn tại trong tự nhiên.
Baker đã công bố
khám phá của mình vào năm 2003. Có lẽ đây là bước đột phá và nhờ đột phá này mà
Baker nhận giải Nobel Hóa học năm 2024. (Đó là tôi đoán mò thế, chưa chắc đã
đúng.)
Một số trong
nhiều loại protein ngoạn mục được tạo ra trong phòng thí nghiệm của Baker được thể
hiện trong hình minh họa dưới đây. Ông cũng đã công bố mã nguồn Rosetta, do đó,
cộng đồng nghiên cứu toàn cầu tiếp tục phát triển phần mềm, tìm ra các lĩnh vực
ứng dụng mới. Tham khảo: https://rosettacommons.org/
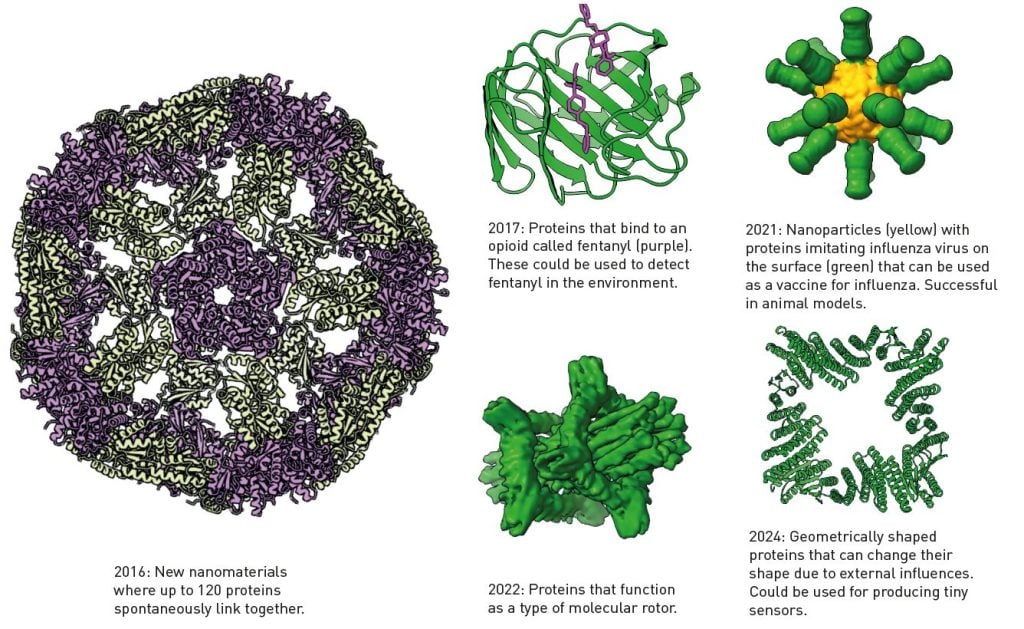 Một số protein được phát triển bằng phần mềm Rosetta của Baker.
Dòng thời gian này
làm nổi bật sự tiến bộ trong lĩnh vực thiết kế protein và công nghệ nano, cho
thấy các protein được thiết kế có thể có những ứng dụng đa dạng, từ phát hiện
thuốc cho đến vắc-xin và cơ chế phân tử. |
-
Tin thêm:
Ngày 5/9/2024,
DeepMind đăng bài AlphaProteo generates novel proteins for
biology and health research
(AlphaProteo tạo ra các protein mới cho việc nghiên cứu y sinh). Theo tôi hiểu
thì AlphaProteo là một mô hình (mạng nơ-ron: neural network) có khả năng
thiết kế các protein mới (giống chức năng của phần mềm Rosetta). Căn cứ theo
bài post thì AlphaProteo có khả năng đặc biệt trong việc thiết kế loại protein
kết dính (binder) với độ ái lực liên kết (binding affinity) cực
cao.
Có thể anh/chị
tò mò đặt câu hỏi: protein kết dính (binding protein) có tác dụng gì? Lấy
một ví dụ cho dễ hiểu. Chúng ta biết rằng SARS-CoV-2 là virus gây ra đại dịch
COVID-19. Để phá vỡ chức năng gây hại của SARS-CoV-2, một trong các phương pháp
là tạo ra các protein kết dính với virus này. Khi có protein kết dính chặt vào
thì chức năng gây hại của SARS-CoV-2 bị vô hiệu hóa (neutralized). Trong
báo cáo sách trắng (whitepaper) của bài đăng, họ đã kiểm thử 4 loại
protein kết dính do AlphaProteo thiết kế tương ứng với 4 biến thể của SARS-CoV-2.
Tất cả 4 loại protein kết dính đó (GDM_SC2BRD_11, GDM_SC2BRD_27, GDM_SC2RBD_104
và GDM_SC2BRD_50) đều vô hiệu hóa thành công các biến thể của SARS-CoV-2.
---
🤔 Suy ngẫm chậm.
🥡 Có
thể dễ dàng nhận thấy Machine Learning đóng vai trò rất lớn trong việc nghiên cứu
y sinh. Tôi đọc nhiều bài báo ca ngợi AlphaFold. Họ ví bước đột phá của
AlphaFold tương tự như Human Genome Project (dự án giải mã gen người), Hubble
Space Telescope (đài thiên văn Hubble) hay việc phát hiện ra hạt Higgs. Machine
Learning giúp giải quyết vấn đề mà cả nửa thế kỷ nay nhân loại gặp bế tắc.
🥡 Có
một xu hướng đang định hình rất rõ ràng: thiết kế, chế tạo các protein de novo.
David Baker có thể coi như tiên phong trong vấn đề này: tạo ra các protein chưa
có tiền lệ, rất độc đáo, có thể ứng dụng vào nhiều lĩnh vực khác nhau trong cuộc
sống. Hẳn nhiên, có nhiều đối tác khác vào cuộc. Machine Learning (như
trường hợp AlphaProteo của Google DeepMind) có thể tạo ra các đột phá mới trong
lĩnh vực này.
Trân trọng
& vui nhã
(_/)
( •_•)
/ >☕
LeVanLoi

Không có nhận xét nào:
Đăng nhận xét